
oben: gemessen, unten: berechnetes Strichspektrum des Schwingungsgrundzustandes.
Für die Zuordnung der K-Quantenzahlen wurde nach Linienpaaren gesucht, welche die berechneten Differenzen zwischen zwei aufeinanderfolgenden K-Komponenten am besten reproduzierten. Diese Differenzen sind erheblich unempfindlicher gegenüber Abweichungen der Rotationskonstanten als die Absolutfrequenzen. Auf diese Art und Weise war schnell etwa ein Dutzend Linien dem Übergang 17<-16 zugeordnet.
Aus diesen Übergangsfrequenzen konnten mittels einer Ausgleichsrechnung neue, korrigierte Rotationskonstanten berechnet werden, die eine genauere Vorhersage weiterer Übergänge erlaubten. Mit diesen neuen gemessenen Übergangsfrequenzen war dann durch eine erneute Anpassung wiederum eine bessere Vorhersage weiterer Übergänge möglich ("bootstrap-Methode"). Zunächst mußten bei der Anpassung die Zentrifugalverzerrungskonstanten noch festgehalten werden. Mit dem Auffinden immer neuer Linien und der genaueren Messung der Übergänge konnten die quartischen Konstanten aber nach und nach mit angepaßt werden.
Die Konstanten, die schließlich aus einer Anpassung von 108 Linien (vgl. Linienliste im Anhang) mit J-Quantenzahlen bis 40 und einer Standardabweichung von 30 kHz erhalten wurden, sind:
| A = 3,332158(13)* GHz | DJ = 0,02149(73) kHz |
| B = 1,19307386(52) GHz | DJK = 1,1008(16) kHz |
| C = 1,08221280(45) GHz | DK = -68,42(32) kHz |
| dJ = 0,00332(16) kHz | |
| k = -0,901 | dK = -2,7513(99) kHz |
| Tabelle E.1: Spektroskopische Konstanten von DMCHDO. *Hier, wie auch bei allen weiteren Angaben, geben Zahlen in Klammern, soweit nicht anders vermerkt, einfache Standardabweichungen der letzten angegebenen Stelle an. | |
Hier ist zu bemerken, daß die gemessenen Rotationskonstanten mit denen, die theoretisch berechnet wurden, sehr gut übereinstimmen. Die Abweichungen liegen für RHF/6-31G* und AM1 bei etwa einem Prozent oder sogar darunter.
|
| Abb. E.1: Ausschnitt aus dem MW-Spektrum von
DMCHDO bei 39 GHz, oben: gemessen, unten: berechnetes Strichspektrum des Schwingungsgrundzustandes. |
Abb. E.1 zeigt einen etwa 170 MHz breiten Ausschnitt des Spektrums, der die oben erwähnte Struktur enthält. Die Modulationsspannung betrug dabei nur etwa 10 V, was einer Stark-Feldstärke von etwa 10 V/cm entspricht. Selbst bei dieser geringen Modulationsfeldstärke waren die Übergänge mit hohen K-Quantenzahlen voll ausmoduliert. Erst bei Übergängen mit K<7 reichten 10 V/cm zum Ausmodulieren nicht mehr aus. Das Strichspektrum am unteren Rand wurde mit den angepaßten Parametern berechnet. Man sieht deutlich, daß viele der Linien nicht zum Spektrum des Moleküls im Schwingungsgrundzustand gehören. Die theoretische Rechnung ergab jedoch, daß das Molekül eine sehr tiefliegende out-of-plane-Knickschwingung besitzt (n37), deren berechnete Frequenz* bei ca. 59 cm-1 liegt. Leider war die Aufnahme des IR-Spektrums (vgl. Abb. E.2) von DMCHDO aus technischen Gründen auf den Bereich oberhalb von ca. 75 cm-1 beschränkt, so daß dieser Wert nicht experimentell nachgeprüft werden konnte. Die Erfahrung zeigt, daß die auf diese Art und Weise ab-initio berechneten Schwingungsfrequenzen meist 10-20% zu hoch sind. So wurden zum Beispiel die Frequenzen der C=C-, bzw. C=O-Streckschwingungen zu 2032 cm-1 bzw. 2094 cm-1 berechnet, jedoch bei 1631 cm-1 bzw. 1665 cm-1 gemessen. Die Frequenz der tiefsten Schwingung könnte also eventuell noch kleiner sein.
* Aufgrund der Größe des Moleküls war eine Schwingungsrechnung mit einem 6-31G*-Basissatz nicht möglich. Der angegebene Wert stammt deshalb aus einer ab-initio-Rechnung mit einem STO-3G-Basissatz.
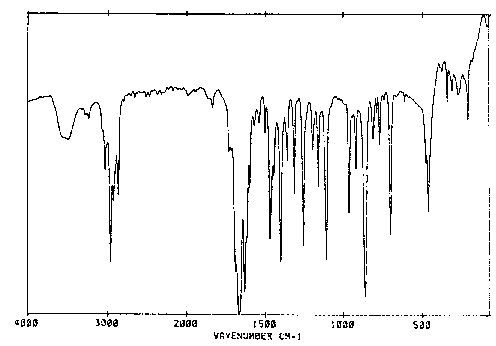 |
| Abb. E.2: IR-Spektrum von DMCHDO. |
Durch gezieltes Suchen nach Linienpaaren mit Abständen, wie sie bereits für den Schwingungsgrundzustand zugeordnet waren, konnte eine Serie von Linien ausgemacht werden, die von einem angeregten Schwingungszustand herrührt. Das Intensitätsverhältnis der Linien des Grundzustandes und dieses angeregten Zustandes ist etwa 3:1, was bei der Meßtemperatur von -30�C einer Schwingungsfrequenz von 185 cm-1 entspricht. Diese Linien gehören also nicht zu der oben angeführten Schwingung, sondern zur nächsthöheren (n36, vgl. Abb. E.3), deren Frequenz mit 233 cm-1 berechnet wurde. Die Skalierung mit dem Faktor 0,8 ergibt hier eine Frequenz von 186 cm-1. Im gemessenen Spektrum findet sich in der Tat eine Bande bei 185 cm-1.
Nach der üblichen Methode konnten nach und nach 29 Linien (vgl. Linienliste im Anhang) diesem Schwingungszustand zugeordnet werden*. Aus den Rotationskonstanten, die aus der Anpassung resultierten, konnten die zugehörigen Schwingungswechselwirkungskonstanten a berechnet werden, mit denen dann versucht wurde, die Linien zu finden, die zum Niveau v36 = 2 dieser Schwingung gehören. Es konnten auch einige Linien gefunden werden, die wohl zu diesem Zustand gehören, jedoch war das Spektrum oft so dicht, daß mehrere Zuordnungen möglich waren. Erschwerend kam hinzu, daß die Rotationskonstanten aufgrund des fast symmetrischen Trägheitsellipsoids des Moleküls stark korreliert waren und mit so wenigen Linien nicht genau genug angepaßt werden konnten, um eine verläßlichere Vorrechnung zu ermöglichen.
* Diese Übergänge wurden jedoch nicht feinvermessen, weshalb die Standardabweichung der Anpassung mit fast 600 kHz auch deutlich höher ist. Für das vorliegende Problem war diese Genauigkeit jedoch ausreichend.
| A = 3,3404(22) GHz | DJ = 0,02149(73) kHz* |
| B = 1,191956(20) GHz | DJK = 1,1008(16) kHz* |
| C = 1,082442(20) GHz | DK = -68,42(32) kHz* |
| k = -0,903 | dJ = 0,00332(16) kHz* |
| dK = -2,7513(99) kHz* | |
| aa36 = -8,2(22) MHz | |
| ab36 = -1,118(20) MHz | |
| ac36 = -0,229(20) MHz | |
| Tabelle E.2: Spektroskopische
Konstanten eines angeregten Schwingungszustandes (v36 = 1) von DMCHDO. *Werte vom Schwingungsgrundzustand übernommen. | |
 |
| Abb. E.3: Schwingung n36 von DMCHDO berechnet bei 223 cm-1, skaliert bei 186 cm-1, gemessen bei 185 cm-1. |
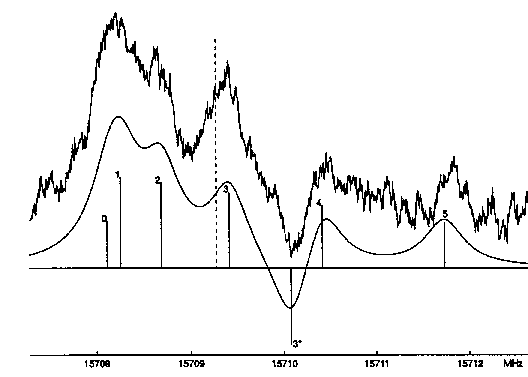 |
| Abb. E.4: Spektrum des Überganges
707 <- 606 von DMCHDO, gemessen bei einem
statischen Feld von 590 V/cm und 825 V/cm Modulation (oben) und berechnet mit Lorentz-Profilen und vollen Halbwertsbreiten von 500 kHz (unten). Die gestrichelte Linie markiert die Nullfeldfrequenz. Die Linienpositionen sind mit ihren jeweiligen M-Werten beschriftet. 3* bezeichnet den Starksatelliten M=3 bei 590 + 825 = 1415 V/cm. Alle anderen M*-Komponenten liegen außerhalbdes dargestellten Bereiches. |
Vor allem im unteren Spannungsbereich, wo die M-Komponenten zum Großteil noch nicht völlig separiert sind, war es nötig, die Einhüllende der Linien anzupassen. Dazu wurde ein Programm entwickelt, das eine Anpassung von bis zu 12 Linien mit Lorentzprofil an das gemessene Signal gestattet (Quellcode im Anhang 3). Die gemessenen Frequenzverschiebungen wurden mit dem Programm RU273 angepaßt. Dabei wurde vorausgesetzt, daß das Dipolmoment vollständig in a-Richtung liegt. Dies wird durch die theoretischen Rechnungen vorausgesagt und nach der hervorragenden Übereinstimmung zwischen gemessenen und berechneten Rotationskonstanten gab es keinen Grund, eine andere Ausrichtung des Dipolmomentes anzunehmen, zumal eine b-Komponente aus Symmetriegründen nicht auftreten kann und eine nennenswerte c-Komponente des Dipolmoments nur für einen erheblich gefalteten Ring zu erwarten wäre.
Der Wert für das Dipolmoment, den die Anpassung lieferte, betrug
m = ma = 4,4522(83) D.
Die Eichung der Zelle erfolgte unmittelbar nach den Messungen an DMCHDO mit OCS, dessen Dipolmoment (0,71521(20) D) sehr genau bekannt ist [42]. Der Wert für die Zellkonstante betrug 1,1794(7) cm-1.
In der Tat wurde bei einer Konformationsanalyse mit zwei molecular-modelling-Programmen (SYBYL [26] und DISCOVER/INSIGHT [27]) ein relativ hoher Wert für die Rotationsbarriere gefunden, nämlich (je nach Geometrie und Methode) zwischen 17 und 20 kJ/mol. Die Aufspaltungen, die bei einer Barriere dieser Höhe zu erwarten wären, liegen für die meisten Übergänge unter 50 kHz, selten auch zwischen 100 und 200 kHz. Bei den verwendeten Drücken (einige hundertstel mbar) betragen die Linienbreiten (volle Halbwertsbreite) allein aufgrund der Druckverbreiterung etwa 300-500 kHz. Eine eventuell vorhandene Aufspaltung durch die innere Rotation würde also im äußersten Fall zu einer Verbreiterung der Linie führen und ist damit nicht meßbar. Dieses Verhalten ist konsistent mit vergleichbaren Dimethyl-Verbindungen. So wurden für 1,1-Dimethylcyclopropan [43a] und 3,3-Dimethylcyclopropen [43b] ebenfalls keine Aufspaltungen aufgrund innerer Rotation beobachtet.
Dieser Effekt könnte allerdings zu einer Ungenauigkeit bei der Frequenzbestimmung führen. Da die Linien aufgrund der inneren Rotation, je nach Übergang, entweder niederfrequent oder hochfrequent verschoben sind, wäre die Folge eine erhöhte Standardabweichung bei der Anpassung. In der Tat ist die Standardabweichung für die Anpassung der Übergänge von DMCHDO mit 30 kHz etwas höher, als zum Beispiel für MCHD, wo diese 23 kHz für die undeuterierte Verbindung, bzw. 18 kHz für die deuterierte Spezies beträgt.
Bei einem planaren Ring weist DMCHDO C2v-Symmetrie auf. Hieraus resultieren 5 Paare von Wasserstoffatomen, die bezüglich einer Drehung um 180 Grad äquivalent sind. Daraus ergibt sich ein Spingewichts-Verhältnis von 33:31. Ein so kleiner Intensitätsunterschied ist bei der verwendeten Methode nicht nachzuweisen, eine Aussage über die Planarität des Ringes kann also auf diese Weise leider nicht getroffen werden.
Auch das Planarmoment Pc allein läßt keine Aussage über die Ebenheit des Ringes zu. Der gemessene Wert von 54,1 u·Å² ist stärker von der Geometrie der Methylgruppen abhängig als von der (Nicht-)Ebenheit des Ringes. So ändert sich das Planarmoment um etwa 1,5%, wenn der Ring um etwa 10 Grad gefaltet wird, jedoch um weit über 3%, wenn die Methyl-Geometrie in sinnvollen Grenzen verändert wird. Die Werte des entsprechenden Planarmoments von dimethylierten Dreiringverbindungen* - leider existieren keine Vergleichsdaten von Sechsringverbindungen - betragen für 3,3-Dimethylcyclopropen 55,3 u·Å² [43b] und für 3,3-Dimethyldiazirin 57,0 u·Å² [43c]. Die Werte sind zwar beide höher als bei DMCHDO, dies läßt sich jedoch leicht durch die, für Dreiringverbindungen typischen 120 Grad-Winkel zwischen den beiden Methyl-Ringkohlenstoff-Bindungen erklären**. So zeigt sich also eine recht gute Übereinstimmung mit Molekü- len, welche einen planaren Ring mit zwei Methylgruppen außerhalb der Ringebene aufweisen: ein erstes experimentelles Anzeichen für einen planaren Ring.
* In diesen Fällen handelt es sich um Pb, da der Ring hier in der (ac)-Ebene liegt.
** Eine ausführliche Behandlung dieser Thematik wird später für das MCHD durchgeführt.
Zur Berechnung eines Pseudo-Trägheitsdefektes müßte die Geometrie der Methylgruppen bekannt sein. Diese wäre jedoch nur durch die aufwendige Präparation von mehreren Isotopomeren (Methyl-13C und Methyl-Deuterium) experimentell zu bestimmen gewesen. Wie bereits oben gezeigt, liefern theoretische Rechnungen jedoch sehr gute Anhaltspunkte für die Geometrie der betrachteten Moleküle. So erschien es sinnvoll, in diesem Fall die theoretisch berechnete Methyl-Geometrie zur Berechnung des Pseudo-Trägheitsdefektes zu benutzen.
Nach Gleichung (25) wurde der Pseudo-Trägheitsdefekt unter Berücksichtigung der c-Koordinaten (die c-Achse steht senkrecht auf der Ringebene) errechnet, wie sie aus der ab-initio-Rechnung mit einem 6-31G*-Basissatz erhalten worden waren:
Koordinate Anzahl Atome |cC| 1,2592 Å 2 |cH| 2,1600 Å 2 |cH'| 1,2787 Å 4Es ergab sich so ein Wert von
Dc = -0,178 u·Å².
Auch wenn dieser Wert aufgrund der verwendeten theoretischen Koordinaten nur eingeschränkte Aussagekraft hat, so bestätigt er doch deutlich eine ebene Struktur. Er entspricht sowohl im Vorzeichen, als auch im Betrag durchaus den Erwartungen, da, wie oben schon erwähnt, eine sehr tiefliegende out-of-planeSchwingung vorliegt. Auch der Vergleich zum 1-Methylen-2,5-cyclohexadien (s.u.) zeigt eine hervorragende Übereinstimmung. Der Wert für die dimethylierte Verbindung liegt etwas höher, was mit der tieferen Schwingungsfrequenz für die "Puckering"-Schwingung gut korreliert. Es darf also mit hoher Wahrscheinlichkeit angenommen werden, daß der Ring im DMCHDO planar ist.
So war also nachgewiesen, daß eine instabile Substanz mit der Molmasse 92 hergestellt worden war. Wie für MCHD zu erwarten war, wurde eine deutliche Instabilität der Verbindung sowohl in der Gasphase, als auch - sehr viel ausgeprägter - in der flüssigen Phase festgestellt. Die beobachteten Mikrowellen-Übergänge waren für Toluol viel zu intensiv. Somit war mit hinreichender Sicherheit anzunehmen, daß es sich bei dieser Substanz tatsächlich um das gewünschte Methylencyclohexadien handelte.
Gajevski und Gortva [6b] geben an, das MCHD gaschromatographisch gereinigt zu haben. Es schien daher die Möglichkeit zu bestehen, die Messungen statisch durchzuführen, was natürlich im Vergleich zu einer Messung im Durchfluß eine erhebliche Substanzersparnis bedeutete. So wurde also die Zelle mit etwa 5 Pa (0,05 mbar) MCHD befüllt und über einen Zeitraum von fast 24 Stunden die Intensität einer bestimmten Linie verfolgt. Eine logarithmische Auftragung (zur Basis 2) ergab eine gute Korrelation bezüglich einer Ausgleichsgeraden (vgl. Abb. E.5). Die Steigung der Geraden entspricht der reziproken Halbwertszeit einer Reaktion erster Ordnung*. Aus den gemessenen Werten ergab sich eine Halbwertszeit t½ von etwa 4 Stunden und 20 Minuten. Es war also möglich, mit zwei Zellfüllungen pro Tag zu messen.
* In [6b] wird für eine benzolische Lösung eine Reaktion zweiter Ordnung beschrieben.
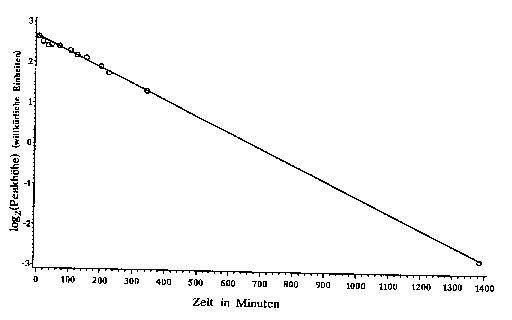 |
| Abb. E.5: Abnahme des Absorptionssignals von MCHD in der Meßzelle. |
Grundlage dieses Verfahrens ist eine Routine, die aus einer Liste von vorgegebenen Linien verschiedene Kombinationen bildet und die vorgegebenen Rotationskonstanten an diese Übergänge anzupassen versucht. Kombinationen, die eine Standardabweichung unter 1 MHz aufweisen, sind dann mögliche Kandidaten für eine richtige Zuordnung. Ein solches Programm war prinzipiell schon vorhanden und es mußten nur noch einige Modifikationen vorgenommen werden.
Bei diesem Verfahren ist natürlich damit zu rechnen, daß es möglicherweise Kombinationen gibt, die zwar - vor allem bei wenigen Linien - eine kleine Standardabweichung liefern, aus denen jedoch Rotationskonstanten resultieren, die nicht mehr sinnvoll sind. Deshalb wurde in das Programm eine Bereichsabfrage eingebaut, die es ermöglicht, eine bestimmte prozentuale Abweichung vorzugeben, um welche die angepaßten Rotationskonstanten von den Startwerten abweichen dürfen.
Zur Bestimmung des Bereiches, in dem mit entsprechenden Übergängen gerechnet werden mußte, wurden die Strukturparameter einzeln um einen kleinen Betrag (bei Abständen 0,1 Å, bei Winkeln 1 Grad) in beide Richtungen verändert und die Auswirkungen auf die drei Rotationskonstanten festgestellt. Dann wurden die Kombinationen gewählt, welche die extremsten Abweichungen verursachen. Mit den so erhaltenen Rotationskonstanten wurden dann Extremspektren errechnet. So ergab sich für jeden Übergang ein Bereich, innerhalb dessen sich die Frequenz mit ziemlicher Sicherheit befinden mußte. Die Linien, welche innerhalb eines solchen Bereiches lagen, wurden in die Eingabedatei für das Anpassprogramm eingetragen.
Mit den so bestimmten "Kandidaten" für die einzelnen Übergänge wurde dann die Anpaßroutine gestartet. Anstatt, wie sonst üblich, für jeden Übergang nur eine Frequenz anzugeben, befanden sich nun im Eingabedatensatz für jeden Übergang alle die Linien, welche im berechneten Bereich lagen. Das Programm bildete dann alle möglichen Kombinationen und gab diejenigen aus, welche eine sinnvolle Anpassung lieferten.
Nach einigen Versuchen, in denen keine Kombinationen gefunden wurden, welche die oben genannten Kriterien erfüllten, wurde dann eine Kombination für vier Übergänge entdeckt, die eine gute Anpassung ergab. Mit den so erhaltenen Rotationskonstanten war es dann möglich, das Spektrum genauer vorzurechnen und weitere Übergänge zu finden. Nach der üblichen "bootstrap-Methode" konnten so schließlich 63 Linien bis J = 60 (vgl. Linienliste im Anhang) mit einer Standardabweichung von 23 kHz angepasst werden. Die erhaltenen Konstanten sind:
| A = 5,1778216(37) GHz | DJ = 0,1384(65) kHz |
| B = 2,6131518(10) GHz | DJK = 0,135(16) kHz |
| C = 1,7558422(09) GHz | DK = 1,074(50) kHz |
| dJ = 0,0440(11) kHz | |
| k = -0,4989 | dK = 0,333(24) kHz |
| Tabelle E.3: Spektroskopische Konstanten von MCHD. | |
Bei der Suche nach geeigneten Übergängen zur Bestimmung des Dipolmoments stellten sich unerwartete Schwierigkeiten ein. Die beiden Übergänge 7(52) <- 6(51) und 8(44) <- 7(43), die zunächst hierfür ausgesucht worden waren, lieferten Dipolmomente, die außerhalb der Fehlergrenzen weit voneinander abwichen. Eine genauere Untersuchung des Termschemas zeigte, daß die Ursache hierfür eine starke Störung der Niveaus des ersten Übergangs war. So liegen zwischen den Niveaus 6(52) und 6(51) nur etwa 3 MHz und auch das zweite Niveau des Übergangs (7(52)) wird durch ein nur 18 MHz höheres (7(53)) gestört. Dies zeigt sich auch in dem maximalen Störkoeffizienten für M=J, der vom Programm RU271 mit ausgegeben wird. Er beträgt für diesen Übergang 0,1. Im Vergleich dazu ist der Wert für den Übergang 8(44) <- 7(43) mit 0,51·10-3 deutlich kleiner. Das Niveau 8(44) ist praktisch ungestört und die nächsten Niveaus zu 7(43) liegen 412 MHz (7(44)), 626 MHz (9(09)) und 635 MHz (9(09)) entfernt.
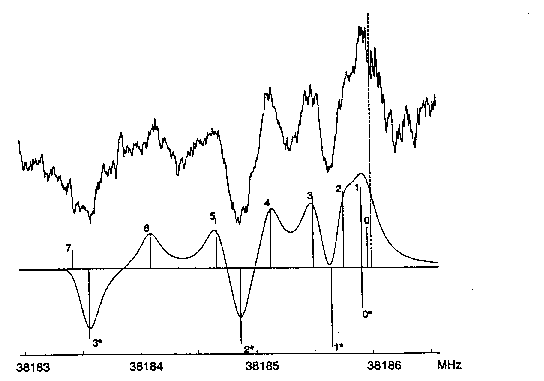 |
| Abb. E.6: Spektrum des Überganges
835 <- 734 von MCHD, gemessen bei einem
statischen Feld von etwa 825 V/cm und 1025 V/cm Modulation (oben) und berechnet mit Lorentz-Profilen und vollen Halbwertsbreiten von 250 kHz (unten). Die gestrichelte Linie markiert die Nullfeldfrequenz. Die Linienpositionen sind mit ihren jeweiligen M-Werten beschriftet. Die mit * bezeichneten Linien stellen Starksatelliten bei 590 + 825 = 1415 V/cm dar. |
Auf der Suche nach einem weiteren Übergang, der die Voraussetzungen für eine einwandfreie Bestimmung des Dipolmoments erfüllt, wurde nur noch ein Übergang gefunden, dessen Niveaus weitgehend ungestört sind und der im Spektrum sauber zu vermessen war, nämlich 8(35) <- 7(34). Hier beträgt der maximale Störkoeffizient nur 0,39·10-4.
Aus den 32 Messungen an M-Komponenten des Übergangs 8(35) <- 7(34) bei Spannungen zwischen 500 und 1800 V (vgl. Abb. E.6 und die Linienliste im Anhang 1) ergab sich ein Dipolmoment von 0,8645(31) D. Der Wert, der aus den 43 Messungen der M-Komponenten des Übergangs 8(44) <- 7(43) erhalten wurde beträgt 0,8434(10). Die Bereiche der einfachen Standardabweichungen überschneiden sich nicht. Deshalb erschien eine gemeinsame Auswertung nicht gerechtfertigt und der Wert des ersten (weniger gestörten) Übergangs wurde als verläßlicher angesehen. Das Dipolmoment von MCHD beträgt also
m = ma = 0,8645(31) D.
Auch nach dieser Meßreihe wurde die Zellkonstante durch Messungen an OCS bestimmt. Sie betrug für diese Messungen 1,1762(9) cm-1.
Eine einfache aber wirkungsvolle Vorgehensweise ist folgende: man bildet das Verhältnis aus den Rotationskonstanten, die sich aus der ab-initio berechneten Struktur ergeben (B(theor.)) und den gemessenen Konstanten (B(exp.)):
fB = B(exp.)/B(theor.) , entsprechend für A und C.
Die so gewonnenen Korrekturfaktoren werden dann mit den Rotationskonstanten multipliziert, die man aus der theoretischen Struktur für das deuterierte Molekül erhält:
B'(korr.) = fB·B'(theor.) , entsprechend für A und C.
Aus den korrigierten Rotationskonstanten für das deuterierte Molekül läßt sich dann ein Erwartungsspektrum errechnen, das mit dem gemessen recht gut übereinstimmen sollte, wenn die berechnete Struktur der r0-Struktur sehr nahe kommt.
In der Tat wurden die Übergänge des deuterierten Moleküls mit weniger als 1 MHz Abweichung von den vorausgesagten Positionen gefunden. Ihre Zuordnung machte so keinerlei Probleme und die Linien konnten sofort feinvermessen werden.
Aus der Anpassung von 44 Linien in bis J = 49 (vgl. Linienliste im Anhang) wurden folgende Konstanten bei einer Standardabweichung von 18 kHz erhalten:
| A = 5,1395303(23) GHz | DJ = 0,1159(78) kHz |
| B = 2,5187678(14) GHz | DJK = 0,128(15) kHz |
| C = 1,7169106(14) GHz | DK = 1,030(53) kHz |
| dJ = 0,04111(68) kHz | |
| k = -0,5314 | dK = 0,273(12) kHz |
| Tabelle E.4: Spektroskopische Konstanten von 4-d-MCHD. | |
Aber im Gegensatz zum DMCHDO ist das Planarmoment, bzw. der Trägheitsdefekt, beim MCHD ziemlich aussagekräftig, da sich nur zwei Wasserstoffatome außerhalb der Ebene befinden, deren Beiträge zum Trägheitsdefekt recht gut abschätzbar sind. Das Planarmoment Pc beträgt beim undeuterierten Molekül
Pc = 1,58785(49) u·Å².
Dies entspricht ziemlich genau dem Wert, den man für ein Paar von Wasserstoffatomen außerhalb der Ringebene erwarten würde. Dies wird durch einen Vergleich mit Molekülen mit vier- und fünfgliedrigen Ringen deutlich, bei denen ebenfalls nur Paare von Wasserstoffatomen außerhalb der Ebene liegen. Eine Zusammenstellung entsprechender Daten enthält Tabelle E.5.
| Molekül | Zahl der Wasserstoff-Paare n | Pc/n (u·Å²) | Literatur |
|---|---|---|---|
 | 2 | 1,643 | [44] |
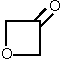 | 2 | 1,639 | [45] |
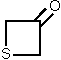 | 2 | 1,699 | [46] |
 | 2 | 1,652 | [47] |
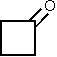 | 3 | 1,663 | [48] |
 | 1 | 1,551 | [49] |
 | 1 | 1,523 | [50] |
 | 1 | 1,520 | [50] |
 | 1 | 1,531 (35Cl) | [51] |
| 1 | 1,583 (37Cl) | [51] | |
 | 1 | 1,588 | diese Arbeit |
| Tabelle E.5: Übersicht über einige Planarmomente pro out-of-plane-Wasserstoffatompaar. | |||
Es ist hier ein deutlicher Unterschied zwischen Molekülen mit Vierringen und solchen mit Fünfringen festzustellen. Das liegt an der veränderten Geometrie der Methylen-Gruppe. Es besteht nämlich eine deutliche Korrelation zwischen dem Ringwinkel (für eine CH2-Gruppe in einem homocyclischen System ist das der C-CH2-C-Winkel) und dem H-C-H-Winkel, bzw. dem C-H-Abstand in der Methylengruppe. Dies wird in den Abbildungen E.7 und E.8 veranschaulicht.
Die Daten für diese Diagramme stammen aus der Datenbank MOGADOC (MOlecular GAsphase DOCumentation) [52]. Sie enthält sowohl bibliographische, als auch molekulare Daten zu molekülphysikalischen Arbeiten in der Gasphase. Eine Suche nach HCH-Winkeln und CH-Abständen in cyclischen Verbindungen ergab 62 Datenpunkte aus 35 Molekülen. Die Beziehungen zwischen dem Ringwinkel a, dem HCH-Winkel b und dem CH-Abstand r wurden mittels linearer Regression aller Datenpunkte erhalten:
b = 127,0(20) - 0,199(21)�a, Korrelation: -0,7689
r = 1,0734(58) + 0.000303(62)�a, Korrelation: 0,5315
=> Pc/n = 2�mH�((1,0734 + 0,000303�a)�sin(127 - 0,199�a)/2)^2Für Vierringe ergibt sich so bei einem Ringwinkel von 90 Grad ein Wert für Pc/n von 1,62 u�Ų. Für Fünfringe (Ringwinkel 108 Grad) erhält man Pc/n = 1,56 u�Ų und für Sechsringe (120 Grad) 1,52 u�Ų. Wenn man diese Werte mit Tabelle E.5 vergleicht, so stellt man fest, daß die experimentellen Werte recht gut mit den berechneten übereinstimmen, jedoch für die Vierringe alle größer sind, was auf die meist sehr tiefliegenden Puckering-Schwingungen zurückzuführen sein dürfte. Bei den sehr starren Fünfringen stimmen die berechneten mit den experimentellen Werten auch gut überein, wobei die Werte eher zu tief liegen. Dies überrascht zunächst etwas, da der Ringwinkel am gesättigten Kohlenstoffatom beim Cyclopentadien zu 103 Grad bestimmt wurde, das Planarmoment also eher noch größer sein sollte. Berücksichtigt man jedoch, daß die in-plane-Schwingungen das Planarmoment verkleinern und dieser Effekt oft (außer für sehr tiefliegende out-of-plane-Schwingungen) größer ist als der entgegengesetzte Effekt der out-of-plane-Schwingungen, so läßt sich auch diese Abweichung erklären.
Für den Sechsring ist der berechnete Wert wiederum etwas zu klein. Hier wirken die Effekte der "Puckering"-Schwingung und des, gegenüber dem regulären Winkel von 120 Grad vermutlich verkleinerten Winkels am gesättigten Methylen-Kohlenstoff zusammen. So entspricht der gemessene Wert für MCHD durchaus den Erwartungen für einen ebenen Ring.
Eine schwache Knickung des Ringes kann jedoch allein auf Grund des Planarmoments nicht ausgeschlossen werden. Der gemessene Wert kann auch bei Diederwinkeln innerhalb des Ringes von etwa 4 Grad gerade noch noch reproduziert werden, wenn man einen C-H-Abstand von 1,09 Å und einen H-C-H-Winkel von 102 Grad annimmt (vgl. Abb. E.9). Da dies jedoch bereits extreme Werte sind, ist eine weitergehende Faltung des Rings, wie dies für das 4,4-dimethylierte Molekül angenommen wurde [10], nahezu unmöglich.
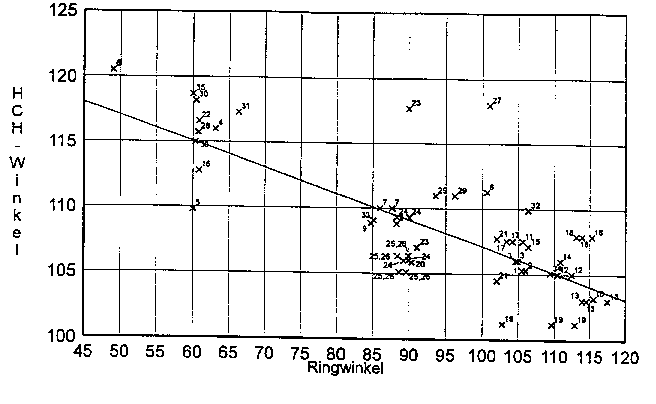 |
| Abb. E.7: Korrelation zwischen Ringwinkel und HCH-Winkel. |
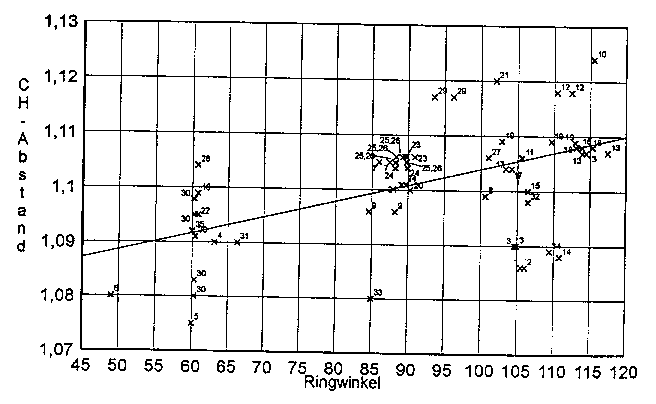 |
| Abb. E.8: Korrelation zwischen Ringwinkel und C-H-Abstand. |
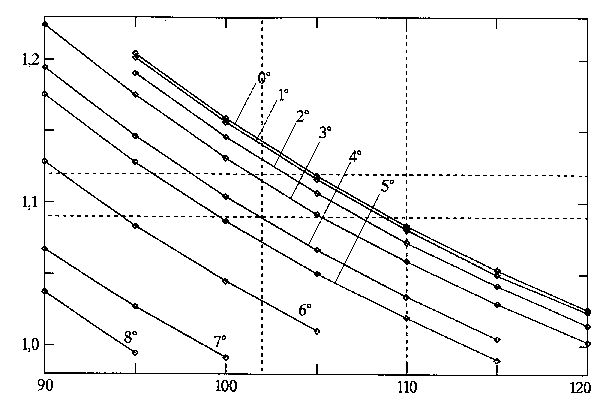 |
| Abb. E.9: Abhängigkeit des H-C4-H-Winkels
und des C4-H-Abstands bei Variation der Diederwinkel (C6-C1-C2-C3 und C2-C3-C4-C5) im Ring. Die Abszisse stellt den H-C4-H-Winkel in Grad dar, die Ordinate den C-H-Abstand in Å. Für neun Diederwinkel (zwischen 0 Grad und 8 Grad) zeigen die Datenpunkte die möglichen Werte für die Methylen-Geometrie, welche das experimentelle Planarmoment Pc reproduzieren. Die gestrichelten Linien markieren Extremwerte, zwischen denen die Parameter erwartungsgemäß liegen sollten. |
Legende zu den Abb. E.7 und E.8:
Die einzelnen Datenpunkte beziehen sich auf folgende Moleküle:
1: N-Chlor-pyrrolidin, axial [53], 2: N-Chlor-pyrrolidin, äquatorial [53], 3: Pyrrolidin [54], 4: Propellan [55], 5: 1,1'-Dimethyl-1,1'-bicyclopropyl [56], 6: 3H-Diazirin [57], 7: 2-Azetidinon [58], 8: 1,1-Difluorsilacyclobutan [59], 9: Azetidin [60], 10: Cycloheptanon [61], 11: Cyclopentenoxid [62], 12: Cyclohexenoxid [62], 13: Cycloheptenoxid [62], 14: Cyclohexanon [63], 15: 2,2-Dihydrofuran [64], 16: Cyclopropylacetylen [65], 17: Cyclopentanon [66], 18: Cyclohepten [67], 19: Bicyclo[3.2.1]octan [68], 20: Cyclobutanon [69], 21: Bicyclo[3.3.0]1.5-octen [70], 22: Cyclopropylsilan [71], 23: 1,1-Dicyanocyclobutan [72], 24: Methylencyclobutan [73], 25: trans-1,1-Bicyclobutyliden [73a, 74], 26: cis-1,1-Bicyclobutyliden [73, 74], 27: 1,3-Dioxolan [75], 28: Cyclopropancarbonsäure, 29: 1,1-Dimethylsilacyclobutan [77], 30: Cyclopropylgerman [78], 31: Dioxiran [79], 32: 2,6,7-Trioxa-1-phosphabicyclo[2.2.1]heptan [80], 33: 1,4,4-Trifluorcyclobuten [81], 34: Hexahydro-1,3,5-trinitro-1,3,5-triazin [82], 35: 1,1-Dichlorcyclopropan [83].
_____________________________________________________________
Aber auch eine geringe Faltung des Ringes kann aus der qualitativen Auswertung der Spektren der deuterierten und der undeuterierten Spezies nahezu ausgeschlossen werden. Dazu sollen im folgenden verschiedene Fälle bezüglich der Potentialfunktion der Knickschwingung des Ringes diskutiert werden (vgl. [46]):
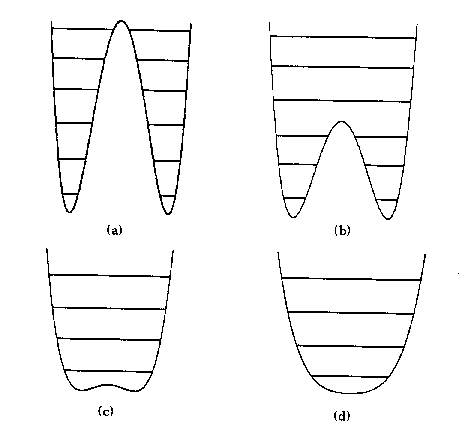 |
| Abb. E.10: Verschiedene Potentialformenfür
die Knickschwingung. Sie haben alle die üblicherweise verwendete Form V = a·x4 - b·x2, wobei x die reduzierte Puckering-Koordinate darstellt. |
Die beiden einzigen mit den gemessen Spektren konsistenten Fälle wären die beiden letztgenannten. In beiden Fällen müßte man jedoch die Struktur des Moleküls im Schwingungsgrundzustand als eben betrachten.
In diesem Fall sollte das Molekül C2v-Symmetrie besitzen, wobei der Ring in der ab-Ebene liegt und die zweizählige Achse, die durch die zwei MethylenKohlenstoffatome definiert wird, die a-Achse darstellt. Das substituierte Wasserstoffatom läge dann in der ac-Ebene und die b-Koordinate sollte exakt Null sein. Mit den Kraitchman-Formeln [20] für einfach substituierte Moleküle ergab sich für die b-Koordinate dieses Wasserstoffatoms ein Wert von 0,060(52) Å1. Es erschien also gerechtfertigt, die Formeln zu benutzen, die für Atome gelten, die in einer Trägheitsebene liegen (dabei wird die b-Koordinate gleichsam als Null vorgegeben). Die so berechneten aund c-Koordinaten waren innerhalb der Fehlergrenzen mit den zuvor erhaltenen identisch. Die genauen Werte für die
Position dieser Wasserstoffatome, welche aus den Kraitchman-Gleichungen erhalten wurden, waren:
ohne Vorgaben H in Trägheitsebene Ungenauigkeit* theor. (6-31G*) |a| 2.5551 2.5550 0.0012 2.538 |b| 0.060 0.0 0.052** 0.0 |c| 0.8673 0.8673 0.0036 0.865Tabelle E.6: Substitutionskoordinaten der 4-Methylen-Wasserstoffatome (in Å). *Nach van Eijck [84]. **Dieser Wert bezieht sich nur auf die Spalte "ohne Vorgaben", da er für die Spalte "H in Trägheitsebene" genau Null ist.
Die Übereinstimmung mit der ab-initio-berechneten Struktur (0,7 bzw. 0,3% Abweichung - der theoretische Wert für |c| liegt sogar innerhalb der experimentellen Fehlergrenzen) ist beeindruckend: ein weiteres Indiz für die Planarität des Ringsystems.
Der Beitrag der beiden "out-of-plane"-Wasserstoffatome zum Planarmoment kann mit der Kenntnis der Koordinaten berechnet werden. Man erhält so den Pseudoträgheitsdefekt. Er beträgt für MCHD:
Dc = 2(2mHc- Pc) = -0,1435(56) uŲ
Dieser Wert ist definitiv zu klein, um irgendeine nennenswerte Nicht-Ebenheit des Ringes zuzulassen. Zwar ist er deutlich negativer, als z.B. beim Cyclopentadienon (0,01617(5) uŲ [35a]), diese Tatsache läßt sich aber ohne weiteres durch eine tiefliegende "out-of-plane"-Schwingung erklären. Die Frequenz dieser tiefsten Schwingung wurde mit 127 cm-1 berechnet (skaliert bei 102 cm-1). Sie sollte einen erheblichen negativen Beitrag zum Pseudoträgheitsdefekt liefern. Auf diesem Hintergrund kann eine nicht-ebene Gleichgewichtsstruktur ausgeschlossen werden.
Diesen eindeutigen Befunden aus der Mikrowellenspektroskopie stehen nur die Ergebnisse der zu Anfang angeführten Elektronenbeugungsarbeit von Trætteberg et al. [10] über 4,4-Dimethyl-1-methylen-2,5-cyclohexadien (DMMCHD) gegenüber. Die Autoren geben für DMMCHD eine Wannen-Struktur an, die innerhalb des Ringes Diederwinkel von etwa 8 Grad aufweist.
Wie bereits im theoretischen Teil angedeutet, tritt bei der Elektronenbeugung prinzipiell ein "shrinkage effect" auf. Da mit der Elektronenbeugung Abstände zwischen den Atomen bestimmt werden, erhält man für ein dreiatomiges Molekül, wie in Abb. E.11, drei Abstände: r1, r2 und R. Für ein starres, nichtschwingendes Molekül gilt dann r1 + r2 = R. Für ein knickschwingendes Molekül erhält man jedoch aufgrund der senkrechten Schwingungsanteile einen kürzeren Abstand R' und das Molekül scheint gewinkelt zu sein. Diesem Effekt kann man durch Einbeziehen der Schwingungsanteile Rechnung tragen. Mit dieser "shrinkage correction" wird dann eine korrekt lineare Struktur erhalten.
 |
| Abb. E.11: Shrinkage-Effekt. |
Trætteberg et al. geben an, eine shrinkage-Korrektur durchgeführt zu haben. Daß die schließlich publizierten Daten aber trotzdem einen gewinkelten Ring beschreiben, ist auf diesem Hintergrund unverständlich. Eventuell rührt diese Diskrepanz daher, daß in derselben Arbeit auch noch ein anderes Molekül, nämlich Bi-(4,4-dimethyl-2,5-cyclohexadien-1-yliden) (Pentaen, XI, vgl. Abb. E.12) untersucht wurde.
 |
| Abb. E.12: Bi-(4,4-dimethyl-2,5-cyclohexadien-1-yliden). |
Da die Beugungsdaten für DMMCHD deutlich schlechter waren, als diejenigen für das Pentaen, wurde die Kraftkonstanten-Analyse für das Pentaen durchgeführt und bei einer Verbesserung diese neuen Parameter für das DMMCHD übernommen. Da im Falle des Pentaens durch die sich gegenüberstehenden aWasserstoffatome, ähnlich wie beim Biphenyl, bei einer ebenen Konformation eine starke sterische Hinderung eintritt, weicht das Molekül dem durch eine Faltung des Ringes aus. Eine Doppel-Wannenstruktur verwundert also nicht. Im Fall des DMMCHD tritt eine solche sterische Hinderung jedoch nicht auf und eine Faltung des Ringes ist aus diesem Grunde nicht nötig. Eine Kopplung der Kraftkonstanten für das Pentaen und das DMMCHD erscheint also zweifelhaft und war im vorliegenden Fall offensichtlich irreführend.
Für das MCHD wurde in dieser Arbeit eindeutig eine ebene Struktur nachgewiesen. Auch im Falle des DMCHDO sprachen alle Fakten für einen ebenen Ring. Ein sterischer Effekt der beiden Methylgruppen in 4-Stellung kann also ausgeschlossen werden. Aus diesen Gründen darf wohl die Interpretation der Beugungsdaten in [10] in Zweifel gezogen werden.
 Zurück zum Inhaltsverzeichnis
Zurück zum Inhaltsverzeichnis
