Alle mikrowellenspektroskopischen Untersuchungen wurden mit einem StarkSpektrometer der Abteilung Chemische Physik durchgeführt. Das verwendete Spektrometer wurde bereits an anderer Stelle ausführlich beschrieben [12, 36], weshalb hier nur ein grober Überblick gegeben werden soll.
Für alle Messungen konnte eine im Temperaturbereich zwischen -70 und +40°C thermostatisierbare 3m-Rechteckzelle aus Messing verwendet werden. Als Strahlungsquellen dienten Rückwärtswellenoszillatoren (18,5-40 GHz) und ein YIG-Oszillator (8-20 GHz).
Übersichtsspektren (Frequenzausschnitt pro Spektrum etwa 50 MHz) wurden
ohne Frequenzstabilisierung aufgenommen, bei der Feinvermessung
(Frequenzausschnitt etwa 2-4 MHz) wurde die Mikrowellenfrequenz über
eine PLL-Schaltung mittels einer FDS-30-Einheit der Firma Schomandl
stabilisiert. Als Referenzfrequenz für die Frequenzmessungen diente ein
digitaler Frequenzgenerator, dessen Genauigkeit gegen das Normsignal des
Langwellensenders Mainflingen der Physikalisch Technischen Bundesanstalt in
Braunschweig geprüft wurde. Die Abweichung war kleiner als
4·10-8, was
bei einer Frequenz von 20 GHz einem Fehler von weniger als 800 Hz
entspricht. Dieser Fehler ist gegenüber den Ungenauigkeiten, die aus
der Linienbreite resultieren, vernachlässigbar. Die typischen
Linienbreiten (volle Halbwertsbreiten) lagen bei der Oxo-Verbindung bei
300-500 kHz und bei der Methylen-Verbindung bei etwa 250 kHz. Daraus
resultieren Ungenauigkeiten bei der Frequenzbestimmung von feinvermessenen
Linien von etwa 20-30 kHz.
Das Detektorsystem des Spektrometers besteht aus einer Diode, deren Ausgangssignal - nach kapazitiver Abtrennung des Gleichspannungsanteils und Verstärkung - phasenempfindlich gleichgerichtet wird. Die dazu nötige Signalmodulation wird über den Stark-Effekt erreicht. Die Stark-Spannung wird von einem Rechteck-Generator erzeugt, der mit einer Frequenz von 33 kHz arbeitet. Das Ausgangssignal des phasenempfindlichen Gleichrichters wird einem Rechner zugeführt, durch den das Verhältnis von Signal zu Rauschen mittels "time-averaging" nochmals deutlich verbessert wird.
Obwohl die verwendete Meßzelle prinzipiell auch im Durchfluß zu betreiben ist, konnte bei allen vermessenen Substanzen statisch gearbeitet werden. Die typischen Drücke in der Zelle betrugen dabei wenige Hundertstel Millibar (2-7 Pa). Je nach Linienstärke waren zum einwandfreien Vermessen der Linien zwischen 100 und 10000 Sweeps nötig. Bei einer maximalen Wiederholfrequenz von knapp unter 10 Hz ergaben sich so Aufnahmezeiten von 10 Sekunden bis zu etwa 20 Minuten.
Zusammen mit den Frequenzmarken wurden die Spektren vom Aufnahmerechner (einem ELTEC-System unter CP/M) auf einen IBM AT überspielt, wo die Frequenzbestimmung komfortabler erfolgen konnte. Von dort wurden die Daten zum Ausdrucken der Spektren und zur Anpassung der gemessenen Linienfrequenzen weiter auf den DEC VAX/VMS-Cluster des Universitäts-Rechenzentrums Ulm übertragen.
Die Anpassung der Daten erfolgte mittels größtenteils vorhandener Programme, insbesondere BC7 (BC2 (Nakagawa), weiterentwickelt von Typke, Lindenmayer und Koch) und MWDOP (Typke) zur Vorausrechnung von Rotationsspektren, ZFAP4 (Typke) zur Ausgleichsrechnung für die gemessenen Rotationsübergänge und RU271 sowie RU273 (beide Rudolph) für die Vorausrechnung und Anpassung der Stark-Effekt-Messungen.
DMCHDO wurde nach dem folgenden, dreistufigen Syntheseplan aus 4,4-Dimethyl-2-cyclohexenon hergestellt (vgl. Abb. D.1):
- Darstellung des Trimethylsilylethers mittels Trimethylsilylchlorid (nach [37]).
- Bromierung unter Spaltung der O-Si-Bindung (nach [38]).
- Eliminierung von Bromwasserstoff mit Chinolin (nach [39]).
Die Darstellung auf diesem Wege liefert ein reineres Produkt als die bereits
beschriebenen einstufigen Synthesen [4c,
4d, 7f, 40a-b]. Es handelt sich dabei um eine Modifikation
der Synthese, die von Plieninger et al. [40c-d] entwickelt wurde und über das Acetat
anstelle des Silylethers verläuft. Der wesentliche Grund für die
Wahl dieses Synthesewegs war, daß es so möglich war, die
Durchführbarkeit einer thermischen HBr-Abspaltung zu prüfen,
welche eventuell einen Zugang zum unmethylierten Cyclohexadienon versprach.
Die thermische Behandlung des Bromids in der Gasphse verlief jedoch nicht
unter HBr-, sondern unter CO-Abspaltung.
 |
| Abb. D.1: Syntheseplan zur Darstellung von
DMCHDO. |
DMCHDO ist eine stabile Substanz und kann unter Luftabschluß längere Zeit gelagert werden, ohne daß eine irgendwie geartete Umsetzung beobachtet werden kann. Deshalb wurde etwa ein Milliliter der Substanz in ein Vorratsgefäß gefüllt, sorgfältig entgast und dann nach Bedarf eine Probe aus diesem Gefäß in die Meßzelle dosiert. Es erwies sich als vorteilhaft, bei einer Temperatur von -30°C und einem Druck von 2-5 Pa (0,02-0,05 mbar) zu arbeiten, jedoch waren die meisten der Linien aufgrund des hohen Dipolmoments so stark, daß eine weitere Optimierung der Aufnahmebedingungen nicht nötig war.
MCHD wurde, wie bereits prinzipiell von Plieninger und Maier-Borst [41] beschrieben, aus Benzoësäure hergestellt. Als letzte Vorstufe diente jedoch nicht das von ihnen verwendete Ammoniumjodid, sondern, wie von Gajewski und Gortva [6b] vorgeschlagen, das entsprechende Aminoxid (vgl. Abb. D.2):
- Birch-Reduktion von Benzoësäure zu 1,4-Dihydrobenzoësäure.
- Umsetzung mit Oxalylchlorid zum entsprechenden Säurechlorid.
- Bildung des Dimethylamids.
- Reduktion zum entsprechenden Amin mit Lithium-Aluminiumhydrid.
- Bildung des Aminoxids mit Wasserstoffperoxid.
- Thermolyse des Aminoxids zu MCHD.
 |
| Abb. D.2: Syntheseplan zur Darstellung von
MCHD. |
Eine filtrierte wässrig-methanolische Lösung von 1,4-Dihydrobenzyl-dimethylaminoxid (X, Darstellung siehe präparativer Anhang) wurde zunächst im Wasserstrahlvakuum, danach etwa eine Stunde bei ca. 1 mbar, eingeengt. Die so erhaltene Masse wurde, ausgehend von Zimmertemperatur, bei langsam bis 80°C steigender Temperatur thermolysiert und die Thermolyseprodukte in einer mit flüssigem Stickstoff gekühlten Falle aufgefangen. Der Vorgang wurde mit einem Quadrupol-Massenspektrometer (MS) beobachtet*.
* Eine massenspektrometrische Unterscheidung zwischen Toluol und MCHD ist nicht möglich. Deswegen wird im folgenden immer Toluol/MCHD für Signale bei m/z = 91-92 verwendet.
Zunächst wurde bei Zimmertemperatur hauptsächlich Toluol/MCHD
erhalten. Dabei trat aber noch kaum Zersetzung ein (fehlende Signale des
zweiten Thermolyseproduktes, Dimethylhydroxylamin (DMHA, m/z=61), welches
einen deutlich kleineren Dampfdruck als Toluol besitzt), so daß es
sich hierbei wahrscheinlich um Toluol aus bereits zuvor zerfallenem Aminoxid
handelte. Bei Erhöhung der Temperatur war dann ein Druckanstieg
bemerkbar und das Spektrum zeigte etwa gleiche Anteile an Toluol/MCHD und
DMHA.
Aus der Kühlfalle, in der die Thermolyseprodukte aufgefangen worden
waren, wurde in eine zweite mit flüssigem Stickstoff gekühlte
Falle umsublimiert. Dazu wurde die Falle mit den Thermolyseprodukten
zunächst aus dem flüssigen Stickstoff in ein Dewargefäß
mit ca. -90°C kaltem Ethanol überführt. Die Temperatur des
Ethanolbades wurde dann durch Zusatz von warmem Ethanol langsam gesteigert.
Auch dieser Vorgang wurde mit dem MS überwacht. Dabei konnte
festgestellt werden, daß sich eine Komponente mit Masse 92 (MCHD oder
Toluol) gut vom DMHA trennen läßt. Bis etwa
-60°C entwickelte sich nur Toluol/MCHD und erst ab ca. -55°C
tauchte DMHA auf, das bei -30°C dominierte. So war
eine effektive, sublimative Abtrennung des gewünschten Produktes
möglich.
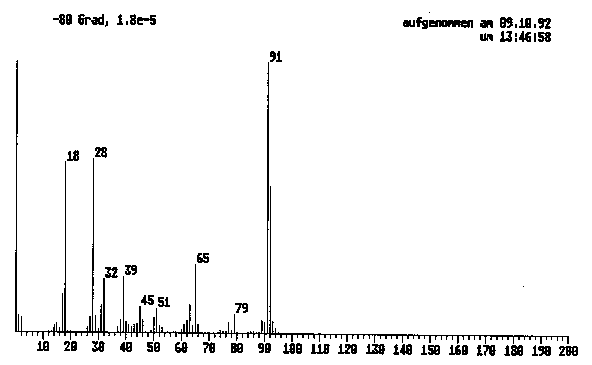 |
| Abb. D.3: Massenspektrum von MCHD. |
 |
| Abb. D.4: Massenspektrum von
Dimethylhydroxylamin. |
Die Dosierung von MCHD erfolgte ähnlich wie die des DMCHDO, jedoch
wurde hier als Vorratsgefäß eine Kühlfalle benutzt, die nur
zum Dosieren kurzzeitig so weit aus dem, mit flüssigem Stickstoff
gekühlten Dewargefäß herausgehoben wurde, daß sich
über dem festen MCHD ein genügender Dampfdruck aufbaute, um die
Zelle zu füllen. Versuche zur Signalintensität bei verschiedenen
Zelltemperaturen ergaben ein Maximum für etwa -30°C (vgl. Abb. D.5). Aufgrund des geringen Dipolmoments wurde, im
Vergleich zum DMCHDO, mit etwas höheren Drücken gemessen,
nämlich mit etwa 8 Pa (0,08 mbar).
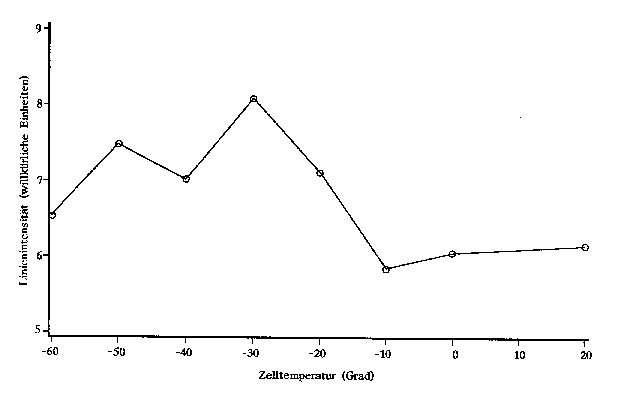 |
| Abb. D.5: Untersuchung über die
Linienintensität von MCHD bezüglich der
Zelltemperatur. |
 Zurück zum Inhaltsverzeichnis
Zurück zum Inhaltsverzeichnis
 Zum vorherigen
Kapitel
Zum vorherigen
Kapitel
 Zum nächsten
Kapitel
Zum nächsten
Kapitel