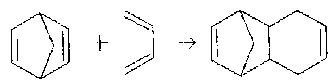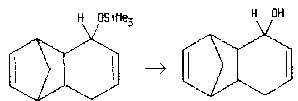Abb.19: Schematischer Aufbau des Mikrowellenspektrometers, nach [19],[20]
Für die Aufnahme von Mikrowellenspektren ist es nun aber nötig, einen bestimmten Frequenzbereich zu überstreichen (Frequenzsweep). Das ist über einen Frequenzmodulationseingang am BWO möglich. Aus Gründen, die später noch dargelegt werden, war es bei dieser Arbeit nicht möglich, die Frequenz wie sonst üblich, sehr langsam zu verändern. Stattdessen mußte der Sweep schnell erfolgen und oft wiederholt werden (Wobbeln). Das wurde dadurch erreicht, daß auf den FM- Eingang eine Sägezahnspannung gelegt wurde, die ganz einfach aus einem Oszilloskop bezogen werden konnte. Dieses Oszilloskop konnte dann auch gleich zur Anzeige von Frequenzmarken oder des Signals verwendet werden, da es genau im Takt mit der Mikrowellenfrequenz läuft. Die Sägezahnspannung wurde noch über einen zusätzlichen Verstärker geführt, mit dem man die Amplitude des Sägezahns (und damit die Sweepbreite) verändern und zusätzlich noch eine variable Sockelspannung (Offset) zum Feinabgleich der Frequenz einstellen kann. Die Triggerung des Oszilloskops erfolgte extern über einen variablen Taktgeber.
Neben der Strahlungsquelle benötigt man auch einige Geräte, die eine genaue Bestimmung der abgestrahlten Frequenz ermöglichen. Als Referenz dient dabei ein quarzstabilisierter Normalfrequenzgenerator, mit dem Frequenzen bis zu 1 GHz erzeugt werden können. Diese Referenzfrequenz und die Mikrowelle werden an einer Diode gemischt. Diese Diode bewirkt zweierlei: erstens erzeugt sie die harmonischen Oberwellen der Referenzfrequenz (Oberwellenkamm) und zweitens bildet sie alle zugehörigen Differenzen und Summen der Oberwellen zur Mikrowellenfrequenz.
Das an dieser Diode anfallende Signal wird über einen Tiefpaß (30 MHz), der als Vorfilter wirkt, einem Radioempfänger zugeführt. Dessen Empfangsfrequenz kann zwischen 1 und 30 MHz eingestellt werden. Von der Diode kann in diesem Bereich nur eine Frequenz kommen, nämlich die Differenz zwischen der Mikrowellenfrequenz und der nächstgelegenen Referenz-Oberwelle (falls diese nahe genug beieinanderliegen, ansonsten erhält man gar kein Signal). Der Radioempfänger erhält also genau dann ein Signal, wenn diese Differenz gleich der eingestellten Empfangsfrequenz ist. Ein typischer Wert hierfür sind 20 MHz.
Abb.20: Der Normalfrequenzgenerator wird so eingestellt, daß die Mikrowellenfrequenz beim Wobbeln immer um
eine harmonische Oberwelle der Referenzfrequenz herum schwankt.
Wenn sich im Laufe eines Frequenzdurchlaufs die Mikrowellenfrequenz von unten her der Referenz-Oberwelle
nähert, so erhält man am Empfänger genau dann ein Signal, wenn die Mikrowelle noch z.B. 20 MHz
entfernt ist. Dann erreicht die Mikrowellenfrequenz den Wert der Referenz-Oberwelle, streicht über diese hinweg und
man erhält schließlich ein weiteres Signal, wenn die Mikrowellenfrequenz um 20 MHz höher ist als die
Oberwelle der Referenz. Welche harmonische Oberwelle zur Differenzbildung beigetragen hat kann man im allgemeinen an
der groben Skala des BWO-Grundgerätes ablesen. Die Mikrowellenstrahlung kann über Hohlleiter und einen variablen Abschwächer in verschiedene
Meßzellen geleitet werden. Verwendet wurden dabei zwei verschiedene Zellen. Für die Referenzmessungen an
Cyclopentadien und Phenol wurde eine 3m-Rechteckzelle verwendet (die Längenangabe bezieht sich auf die
Modulationsstrecke). Die Stark-Elektrode ist hierbei in Form einer rechteckigen Platte über fast die gesamte
Zellenlänge realisiert, die an den schmalen Seiten der Zelle in je einer Längsrille einer Teflonisolierung ruht.
Um die Transmission möglichst wenig zu stören sind die Ein- und Auslaßöffnungen als schmale
Längsschlitze in der breiten Oberseite ausgeführt. Dadurch erhält man zwar sehr gute
Transmissionseigenschaften, jedoch ist ein schnelles Durchströmen, wie es für die Messung instabiler
Substanzen nötig ist, dabei nicht möglich. Andererseits kann man in einer Rechteckzelle keine so großen
Öffnungen anbringen, wie sie für ein schnelles Durchströmen benötigt würden, ohne die
Transmission in unvertretbarer Weise zu stören.
Hierfür wurde dann eine Rundzelle benutzt. Sie weist größere Ein- und Auslaßöffnungen
auf (Nennweite 25 mm), wodurch ein wesentlich rascheres Durchströmen und damit eine kürzere Verweilzeit
der Substanz in der Zelle erreicht wird. Zusätzlich befindet sich im Einlaß ein kleiner Glasfinger, der das
senkrecht zur Zelle einströmende Gas parallel zur Zelle umlenkt. So wird die Strömung noch etwas besser
unterstützt. Die Öffnung dieses Glasfingers besitzt einen Durchmesser von etwa 8 mm und stellt so, neben dem
Auslaßventil, die engste Stelle im Durchflußsystem dar.
Bei der Rundzelle besteht die Stark-Elektrode aus einem eineinhalb Meter langen Metallrohr, das an den Enden
kegelförmig abgeschlossen ist. Das Rohr wird durch sechs Teflonstäbchen in der Zelle zentriert.
Bei früheren Arbeiten in unserer Abteilung zeigte sich, daß Metalloberflächen bei gewissen
Substanzen die Zersetzung katalysieren können. Deshalb sind sämtliche Oberflächen der Rundzelle mit
einer dünnen Teflonschicht überzogen, um so jeden Metallkontakt zu vermeiden.
Aus der Zelle abgesaugt wird die Probe über mehrere Kühlfallen durch einen Pumpstand, der aus einer
Diffusionspumpe und, als Vorpumpe, einer Drehschieberpumpe besteht.
Zusätzlich zum Mikrowellenspektrometer wurde zwischen Zellausgang und Pumpstand eine Verzweigung zu
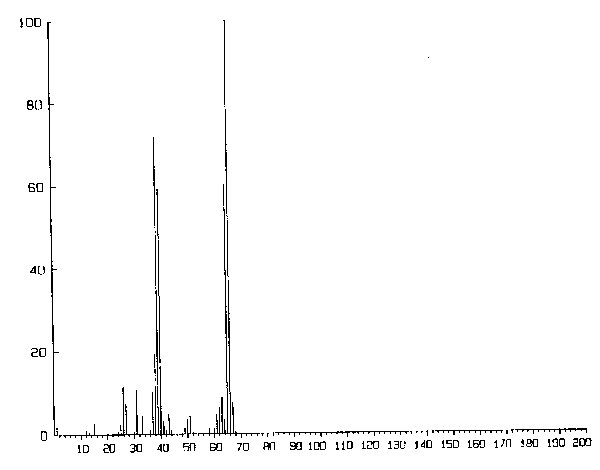
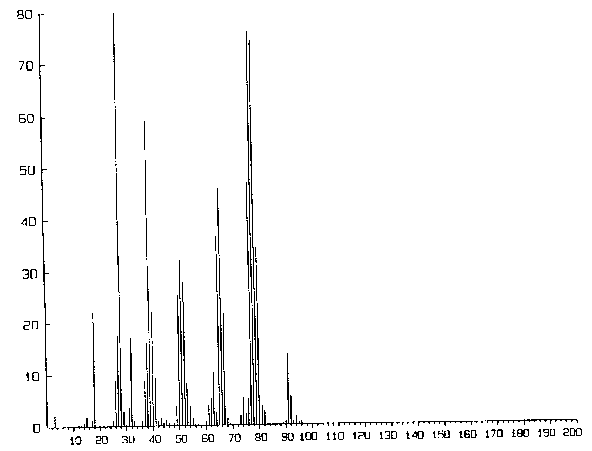
einem Quadrupol-Massenspektrometer (bis 200 AME) vorgenommen, um die Zersetzungsprodukte massenspektrometrisch
nachzuweisen. Die Detektion bereitet bei der Rotationsspektroskopie aufgrund der geringen Absorption ziemliche Schwierigkeiten. Man
darf nicht vergessen, daß im Gegensatz zu anderen Absorptionsmessungen wie UV- oder IR-Spektroskopie, wo der
Anteil der absorbierten Strahlung im Prozent-Bereich liegt, bei der Mikrowellenspektroskopie im ppm-Bereich und darunter
gearbeitet wird. Aus diesem Grund ist der Detektionsteil des Spektrometers die komplizierteste Komponente.
Als eigentlicher Detektor der Mikrowellenstrahlung dient eine Spitzen-Diode, die in einem Hohlleiter-Anpassglied
untergebracht ist. Bei sehr starken Absorptionen ist es möglich an der Dioden-Ausgangsspannung direkt die
Transmissionsverringerung zu erkennen. Im Normalfall ist diese Verringerung jedoch so klein, daß sie im Rauschen
vollständig untergeht. Die ganze Technik, die im Detektorteil Anwendung findet, dient also nur dazu, das
Verhältnis zwischen Signal und Rauschen zu verbessern. Ein wichtiges Hilfsmittel ist dabei die Modulation des Absorptionssignales. Da die Rotationsspektroskopie nur an
Molekülen mit permanentem elektrischem Dipolmoment möglich ist, bietet sich hier die Ausnutzung
elektrischer Felder - also des Stark-Effektes an. Die einzelnen J-Niveaus sind nämlich bezüglich ihrer M-
Quantenzahl (also der räumlichen Ausrichtung des Drehimpulses) entartet. Diese Entartung wird durch ein
äußeres elektrisches Feld teilweise aufgehoben, da nun nicht mehr alle räumlichen Ausrichtungen des
Drehimpulses zum äußeren Feld energetisch gleichwertig sind. Deshalb wird der Übergang i.a. in mehrere
Komponenten aufgespalten.
Abb.21: An der Diode liegt (mit 33 kHz moduliert- hier allerdings sehr vereinfacht dargestellt) sowohl das
Absorptionssignal ohne Feld (Mitte rechts), als auch das mit Feld (links ganz oben) an, allerdings fällt das rechte
Signal logischerweise nur in den Halbperioden mit ausgeschaltetem Feld an (rechts unten), das linke nur in den Halbperioden
mit angeschaltetem Feld (links unten).
Bei angelegtem Feld tritt der Übergang also nicht mehr an derselben Stelle auf wie ohne Feld. Schaltet man
nun das Feld periodisch an und aus (hier realisiert als Rechteckmodulation mit einer Frequenz von 33 kHz), so wird ein
bestimmter Übergang an einer bestimmten Stelle nur dann zu sehen sein, wenn das Feld aus ist, an einer anderen nur
dann, wenn es angelegt ist; das Absorptionssignal ist also mit der Frequenz der Starkspannung moduliert. Nach Abkoppeln des Gleichspannungsanteils kann man das Diodensignal (33 kHz) phasenempfindlich gleichrichten.
Dazu benutzt man einen Lock-In-Verstärker, der seine Referenzfrequenz aus dem Stark-Generator bezieht. So besteht
zwischen Signalträger und Referenz immer eine feste Phasenbeziehung. Über einen Phasenschieber am Lock-In-
Verstärker können schaltungsbedingte Phasenverschiebungen ausgeglichen werden. Stellt man die Phasenlage
so ein, daß die Phasendifferenz zwischen Referenz und Signal genau 0° oder 180° beträgt, so
erhält man maximale Signalintensitäten. Dabei wird aber, da das Signal bei eingeschaltetem Feld
gegenüber dem Signal ohne Feld genau um 180° verschoben ist, die Diodenspannung bei angelegtem Feld mit
entgegengesetztem Vorzeichen verstärkt. Das ist der Grund, weshalb die Stark-Satelliten im Spektrum nach unten
zeigen, während die Linien ohne Feld nach oben gerichtet sind.
Der wichtigste Vorteil des Lock-In-Verstärkers besteht darin, daß, auf Grund der festen Phasenbeziehung
zur Referenz, die Bandbreite des Verstärkers nur durch den nachgeschalteten Tiefpaß bestimmt wird. Deshalb
können vom Rauschen nur die Anteile passieren, deren Frequenzen nahe bei der Modulatiosfrequenz liegen. Alle
anderen Anteile werden so praktisch weggefiltert. Dadurch steigt das Verhältnis von Signal zu Rauschen
beträchtlich. Um das Signal nun noch mehr vom Rauschen zu befreien wäre es möglich, mit einem Tiefpass geringer
Bandbreite zu arbeiten und dadurch die meist schnellen Amplitudenschwankungen des Rauschens noch besser auszufiltern.
Dabei müßte man dann aber sehr langsam über die Absortionslinie hinwegfahren, um
höherfrequente Fourierkomponenten im Signal zu vermeiden, die vom Tiefpaß weggefiltert würden.
Dadurch erhielte man ein verzerrtes Signal. Ein so langsamer Frequenzvorschub erweist sich für instabile
Moleküle jedoch als unbrauchbar, da meist nicht garantiert werden kann, daß die Verhältnisse über
den gesamten Sweep hinweg konstant bleiben. Eine Konzentrationsänderung in der Zelle könnte so eine
Absorptionsänderung vortäuschen, die gar nicht vorhanden ist.
Einen Ausweg aus diesem Problem bietet das Time-averaging-Verfahren, das erst durch den Einsatz von schnellen
elektronischen Rechnern ermöglicht und ursprünglich für NMR- und ESR-Anwendungen entwickelt
wurde. Man schließt dabei einen Kompromiß. Man macht den Frequenzvorschub so schnell, wie es nötig
ist, um sicher konstante Verhältnisse über einen gesamten Sweep hinweg zu haben (in unserem Fall etwa 0,1
Sekunden). Man muß dann mit einer entsprechend erhöhten Bandbreite arbeiten. Das dadurch bedingte
größere Rauschen beseitigt man nun andererseits aber dadurch, daß man den Sweep öfters
wiederholt (deshalb der oben schon erwähnte Sägezahn als Frequenzmodulation) und die
Signalintensitäten mit einem Rechner aufaddiert. Es läßt sich mathematisch zeigen, daß dabei das
(statistische) Rauschen nur mit der Wurzel der Anzahl der Sweeps anwächst, während das (kohärente)
Absorptionssignal sich linear aufaddiert. Man erreicht bei 100 Sweeps also eine Verbesserung des Verhältnisses
zwischen Signal und Rauschen um einen Faktor Zehn.
Es ist dabei allerdings wichtig, daß jeweils die richtigen Abschnitte des Spektrums aufaddiert werden, das
heißt, daß in einen bestimmten Kanal im Rechner immer das Signal bei derselben Mikrowellenfrequenz
gespeichert wird. Da der Sägezahn des Oszilloskops, der den Frequenzvorschub steuert, hinreichend stabil ist,
muß nur noch dafür gesorgt werden, daß der Rechner immer bei der selben Frequenz mit seiner Arbeit
beginnt.
Das wird dadurch erreicht, daß ein zweiter Radioempfänger an die Mischerdiode angeschlossen ist, dessen
Frequenz (meist 21 MHz) so eingestellt wird, daß seine Frequenzmarke erzeugt wird, kurz bevor die
Mikrowellenfrequenz den für den linken Rand des Frequenzfensters gewünschten Wert erreicht. Diese Marke
wird dem Rechner als Triggermarke zugeführt. Um zu verhindern, daß der Rechner schon beim
Rücksprung des Sägezahns oder bei der zweiten Marke (21 MHz über der Referenz-Oberwelle) getriggert
wird, läuft das Triggersignal erst noch durch eine Torschaltung, die vom Taktgeber beim Startimpuls geöffnet
wird und sich nach einer gewissen einstellbaren Zeit wieder schließt.
Als Rechner für die Aufaddierung der Meßwerte diente ein unter CP/M betriebener ELTEC E3-182 mit
einem in unserer Abteilung entwickelten Programm.
Abb. 22: Nachdem der vorherige Sweep mit dem Rücksprung beendet wurde, wartet das Oszilloskop auf den
neuen Triggerimpuls vom Taktgeber. Ist dieser erfolgt, beginnt der neue Sweep. Dabei ist das Tor offen, durch das der
Rechner seinen Triggerimpuls bekommt. Nach der eingestellten Zeit schließt sich das Tor und der Rechner bleibt bis
zum nächsten Sweep von Triggerimpulsen unbehelligt. Wenn sowohl die Daten des Spektrums als auch die Frequenzmarken im Rechner gespeichert sind, werden sie mit einem
Datenübertragungsprogramm zur Auswertung an einen zweiten Rechner geschickt. Prinzipiell ist das nicht
nötig, da auch für den ELTEC ein Auswerteprogramm existiert, aber da der zweite Rechner (ein IBM-AT) die
Daten graphisch besser auswerten kann und die Auswertung parallel zu einer weiteren Messung erfolgen kann wurde dieser
Weg beschritten. Die IR-Spektren wurden mit einem Infrared Grating Spektrometer 457 der Firma Perkin-Elmer aufgenommen. Film-
Spektren wurden mit CsI-Fenstern, Festkörperspektren mit KBr-Preßlingen durchgeführt.
Die Kernresonanz-Spektren wurden zum Teil von der Sektion Kernresonanz- Spektroskopie mit einem Bruker MSL 300
Fourier-Transform-Spektrometer angefertigt. Der andere Teil der Massenspektren wurde in der Abteilung Organische
Chemie I an einem Varian EM 360L-Spektrometer aufgenommen. Die Substanzen wurden dazu in
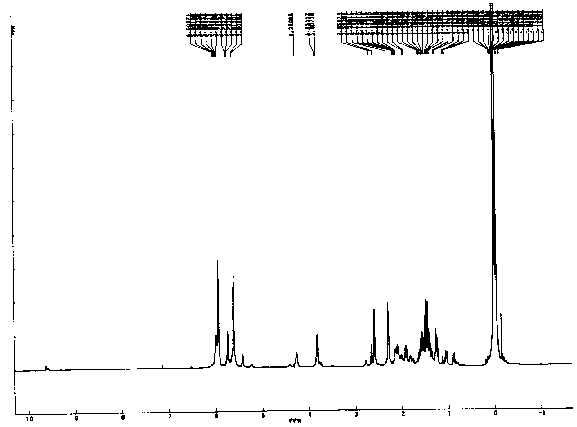
CDCl3 gelöst. Als interner Standard diente Tetramethylsilan (TMS).
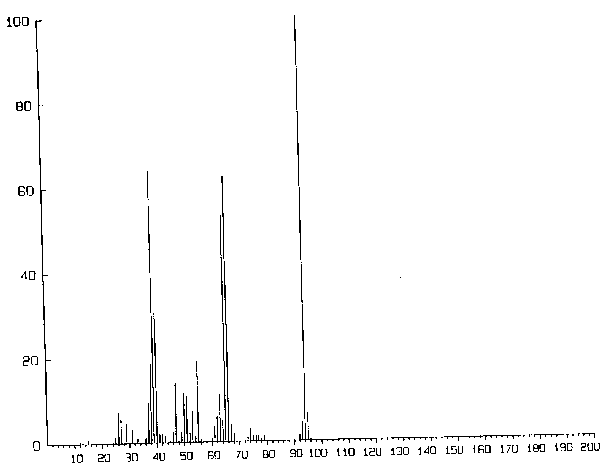
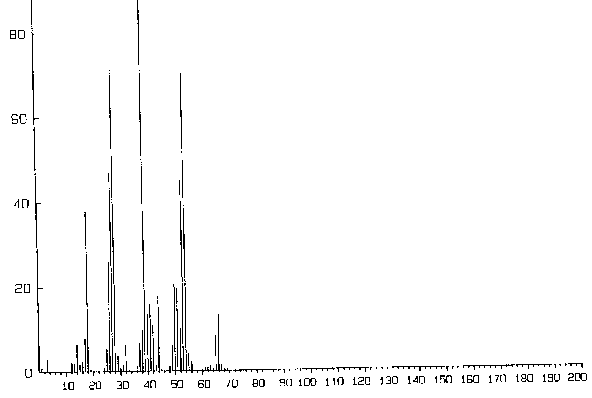
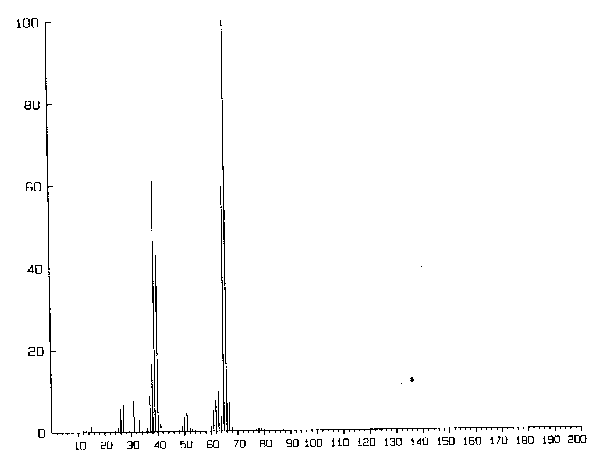
Die Massenspektren des präparativen Teils wurden von der Sektion Massenspektrometrie an einem Varian MAT
711 aufgenommen. Die Spektren, die im Laufe der mikrowellenspektroskopischen Arbeit angefertigt wurden, stammen von
einem Ametek/Dycor Quadrupol-Restgasanalysator. Ausgegeben wurden die Spektren auf einem Laserdrucker des
Universitäts-Rechenzentrums Ulm über ein, im Laufe dieser Arbeit entwickeltes, Fortran-Programm auf einer
VAX 6340 der Digital Equipment Company.
Die verwendeten Chemikalien wurden von den Firmen Aldrich, Merck und Fluka bezogen.
Alle empfindlichen Substanzen, insbesondere die Zwischenstufen, wurden bis zur Verwendung bei ca. -40°C
gelagert.
In den Präparationsvorschriften wird folgende Nomenklatur verwendet:
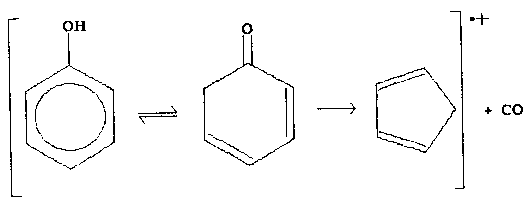
Der Grundkörper (1,4-Methano-hexahydronaphthalin) kann als formales Additionsprodukt aus Cyclopentadien
(CP) und Cyclohexadien (CH) aufgefaßt werden und wird kurz als CPCH bezeichnet. Die davon abgeleiteten
Verbindungen tragen dann Vor- oder Nachsilben, die die Substituenten anzeigen. So steht TMSO- für die
Trimethylsilyloxy-Gruppe, -ol für eine Hydroxy-Gruppe und -on für eine Keto-Gruppe. Shiner, Vorndam und Kass veröffentlichten zwar den groben Reaktionsweg, über den sie zu der
gewünschten Vorläuferverbindung kamen, schilderten jedoch nicht die genauen Bedingungen. Diese Unterlagen
wurden angefordert, in der Zwischenzeit jedoch schon ein Versuch gestartet, die Synthese nachzuvollziehen.
Dazu wurden 4 ml Trimethylsilyloxybutadien (22,8 mmol) mit 6 ml Norbornadien (55,6 mmol) in Toluol gelöst
und etwa 16 Stunden unter Rückfluß gekocht (über Nacht mußte aus Sicherheitsgründen die
Heizung abgestellt werden). Der Verlauf der Reaktion wurde mit IR-Spektren verfolgt. In dieser Zeit wurden keine
signifikanten Änderungen beobachtet. Die Reaktionsmischung wurde deshalb weitere 12 Stunden auf etwa 50 Grad
gehalten. Auch nach dieser Zeit war im IR-Spektrum keine Änderung zu erkennen. Die folgende Destillation erbrachte
nur die Ausgangsprodukte und einen gelben, äußerst hochsiedenden, zähen Sumpf, der nicht weiter
aufgearbeitet werden konnte und auch mengenmäßig unbedeutend war.
Es erschien deshalb ratsam, auf das Eintreffen der Präparationsvorschriften von Shiner, Vorndam und Kass zu
warten und währenddessen einen alternativen Weg zu beschreiten, nämlich die Darstellung über den
reinen Kohlenwasserstoff (CPCH), die auch von Lasne, Ripoll und Denis gewählt wurde. In einen Autoklaven, der mit 162 ml Norbornadien (132 g 1,8 mol) und einer Spatelspitze Hydrochinon befüllt
war, wurden 52 ml Butadien (32,5 g 0,6 mol) aus einer Kühlfalle einkondensiert. Die Mischung wurde 30 Stunden auf
ca. 140 Grad geheizt. Bei der anschließenden Destillation ging zunächst unverbrauchtes Norbornadien
über (90°C). Nachdem dieses abdestilliert war, wurde im Vakuum weiterdestilliert. Bei 90°C und 16 mbar
wurde so eine Rohfraktion von CPCH erhalten. Die anschließende Rektifikation über eine vakuumisolierte
30cm-Vigreux-Kolonne lieferte 37,9 g (0,26 mol = 43% d.Th.) CPCH (nD20 = 1,5148).
5.2 Die Absorptionszelle
5.3 Detektion
5.3.1 Starkmodulation

5.3.2 Phasenempfindliche Gleichrichtung
5.3.3 Time-Averaging

5.4 Auswertung der Daten
6 Präparativer Teil
6.1 Erster Versuch zur Darstellung von TMSO-CPCH (V: 1,4-Methano- 5-trimethylsilyloxy-
1,4,5,8,9,10-hexahydronaphthalin)

6.2 Darstellung von CPCH (III: 1,4-Methano-1,4,5,8,9,10-hexahydronaphthalin) (nach [6])