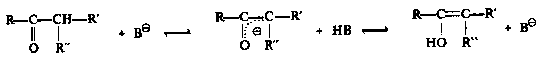Abb.3a: Imin-Enamin-Tautomerie: 1-Amino-cyclohexen nach [31].
Abb.3b: Nitro-Aci-Nitro-Tautomerie: p-Nitrophenyl-nitromethan nach [32].
Abb.3c: Nitroso-Oxim-Tautomerie: p-Benzochinon-monoxim nach [32].
Bei der Umwandlung von Cyclohexadienon in Phenol handelt es sich um einen Fall von Keto-Enol-Tautomerie,
weshalb hier etwas näher auf diese Form der Tautomerie eingegangen werden soll. Geprägt wurde der Begriff der Tautomerie von Butlerow (1877) und Laar (1885). Eine sehr frühe
Bedeutung in der Literatur fand der von Geuther 1863 entdeckte Acetessigester. Zwischen Geuther und Frankland entbrannte
ein heftiger Streit über die richtige Strukturformel für diese Verbindung. Erst Knorr konnte dann 1911 zeigen,
daß hier ein Fall von Tautomerie vorliegt und so eigentlich beide recht hatten.
Abb.4: Acetessigester. (a) Keto-Form, (b) Enol-Form
Die Keto-Enol-Tautomerie spielt vor allem bei den Reaktionen von Carbonylverbindungen eine bedeutende Rolle.
Der erste Mechanismus, der eine Enolisierung des Ketons als geschwindigkeitsbestimmenden Schritt beinhaltete, wurde 1904
von Lapworth [23] für die Halogenierung von Ketonen vorgeschlagen.
Seitdem sind viele tausend Veröffentlichungen zu diesem Thema erfolgt und Toullec [12] bezeichnet die Enolisierung gar als 'einen der am besten dokumentierten Prozesse in der organischen Chemie'.
Die Mechanismen der Enolisierung im speziellen und der Tautomerisierung im allgemeinen sind folgende: Eine Base deprotoniert das Keton am a-Kohlenstoff (CH-Acidität) und bildet so das Enolat-Anion, welches
anschließend wieder protoniert wird. Die Lage des Gleichgewichtes zwischen Enol und Keton wird nun ganz
entscheidend davon bestimmt, an welcher Stelle die Protonierung leichter erfolgt, oder mit anderen Worten: welches Zentrum
basischer ist.


2.3 Geschichte und Bedeutung der Keto-Enol-Tautomerie

2.4 Enolisierungsmechanismen und Gleichgewichtslage
2.4.1 Basenkatalyse