
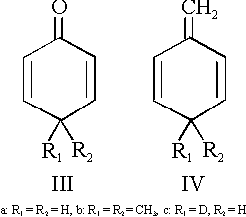
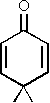
Cross-conjugated compounds with six-membered rings are of special interest since they represent tautomeric forms of aromatic counterparts. The oxo-compound 2,5-cyclohexadienone (IIIa) is a keto-tautomer of phenol. This is also the reason of its high instability which has made it impossible up to now to study this molecule by microwave spectroscopy. We were, however, able to record and assign the spectrum of the corresponding dimethyl-compound (IIIb) [3].
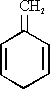
In this paper we wish to present our investigations on the methylene compound 1-methylene-2,5-cyclohexadiene* (IVa) - a tautomer of toluene. An electron diffraction paper [4] on 4,4-dimethyl-1-methylene-2,5-cyclohexadiene (IVb) claims a non-planar ring structure with dihedral angles within the ring of about 8 degrees. One of our aims in this work was to check this by microwave spectroscopy.
*The correct nomenclature for (IVa) would be 3-methylene-1,4-cyclohexadiene. However, for easier reference to various substituted cyclohexadienes we will use "2,5-cyclohexadiene" as the basic unit. The unsaturated exocyclic group is referred to as position 1, the saturated carbon atom on the opposite side of the ring is referred to as position 4.
Unless denoted otherwise whenever a value is given with error limits (in parentheses) these denote the standard deviation or the uncertainty resulting from error propagation in units of the last digits given.
Preparation
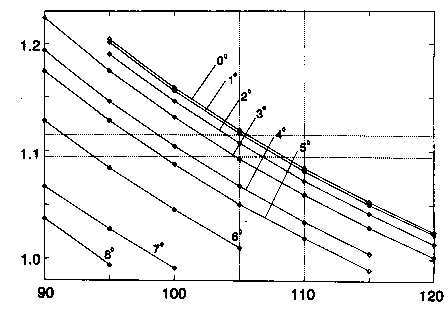
1-Methylene-2,5-cyclohexadiene (IVa) has been prepared following the method of Plieninger and Maier-Borst [5]
with the modification proposed by Gajevski and Gortva [6]:
First benzoic acid (V) is hydrogenated in a Birch reduction [7]. The dihydrobenzoic acid (VI) is then converted to the corresponding amine by standard procedures: formation of the acid chloride (VII) with oxalyl chloride [5]; the acid chloride is allowed to react with dimethylamine to give the dimethylamide (VIII) [5] which is in turn reduced to the amine (IX) with LiAlH4 [5]. The amine is oxidized with H2O2 according to Cope et al. [8]. Thermolysis of the amine oxide (X) finally gives methylenecyclohexadiene (IVa) and dimethylhydroxylamine [6].
The thermolysis was monitored by a quadrupole mass spectrometer. Thus it could be observed that even at
temperatures barely above room temperature a noticeable amount of the amine oxide decomposes. The main part
was, however, thermolyzed at about 65°C. The products were collected in a trap cooled with liquid nitrogen. From
this trap the pyrolysate could easily be separated by fractional sublimation. The most volatile compound was the
desired hydrocarbon. For the microwave measurements methylenecyclohexadiene could thus be passed directly
from the original cooling trap into the cell.
As long as the contents of the trap were kept cool enough so as to prevent melting of the contents, the hydrocarbon
could be handled quite easily and no significant decomposition, oligomerization or tautomerization could be
observed. However as soon as the trap was warmed enough to melt the contents, practically no
methylenecyclohexadiene could be recovered.
Microwave Spectrum and assignment
The measurements were carried out at -30°C and about 3 Pa (0.03 mbar) using a conventional Stark spectrometer
with a 3m cell. The compound was found to be sufficiently stable for static measurements. For the aforementioned
conditions we determined a half life within the cell of about 4 hours. Ab initio and semi-empirical calculations were
very helpful for first guesses of the rotational constants. It turned out that the calculated constants deviated from the
experimentally determined ones by as few as some tenths of a percent. This is in agreement with the experiences
from 4,4-dimethyl-2,5-cyclohexadienone (IIIb) [3].
Due to the relatively small dipole moment first trials of assignments were made in the region between 35 and 40 GHz where the most intense transitions could be expected. Since a powerful computer (a DEC VAX 9000-410 of the computational center of the University of Ulm) was available we tried an "automatic assignment". First we determined reasonable ranges for each of the rotational constants by investigating the derivatives of these constants with respect to the different molecular parameters. Using these ranges it was possible to calculate ranges for some intense low-J-transitions for which the contributions of centrifugal distortion should be below 1 MHz. Now all lines found in these ranges were fed into a computer program, which succesively fitted each possible combination to a rigid rotor model. Combinations resulting in a standard deviation of less than 1.5 MHz and leaving the rotational constants within the given ranges were printed. After some unfruitful trials we found the correct assignment for four transitions. The rotational constants that resulted from this fit were accurate enough to assign many other transitions within a relatively short period of time.
As expected the spectrum followed a-type selection rules and we were able to assign 63 transitions between 12 and 40 GHz with a maximum J of 60 (cf. Table 1). The least squares fit showed a standard deviation of 23 kHz and yielded the three rotational constants and all five quartic centrifugal distortion constants (see Table 2).
The dipole moment was determined from a least squares fit of 32 measurements of Stark shifts of the M components of the transition 8(35)-7(34) and was found to be 0.8645(31) D along the a axis. The cell was calibrated using the dipole moment of OCS [9]. The figure below shows a sample spectrum of the transition 8(35)- 7(34) along with a calculated spectrum.
Spectrum of the transition 8(35)-7(34) recorded at a static field of 825 V/cm with a modulation field of 1025 V/cm (above) and calculated
using Lorentzian line-shapes with line-widths (FWHM) of 250 kHz (below). The zero-field frequency of the transition is shown (---). The signals
are labeled with their respective values of M. The signals marked with an asterisk represent the Stark lobes.
As mentioned earlier, one of our aims was to find out whether the ring of this molecule is planar or not. When the
ring is planar the molecule possesses C2v symmetry. This would result in four pairs of identical hydrogens which
would lead to a spin weight ratio of 17:15. However such a small difference could not be ascertained with the
technique used.
The planar moment Pc was found to be 1.58785(49) amuŲ (using the conversion factor 505379.1 amuŲMHz).
This is what would be expected for a planar ring with one pair of hydrogens out of the plane. This value is also in
good agreement with the values found for planar four-membered and five-membered rings with only pairs of
hydrogen atoms out of the plane (cf. Table 3). The methylene geometry should be
suspected to change systematically when moving from four- to six-membered rings. As the C-C-C ring angle
increases the H-C-H angle gets smaller, thus reducing the planar moment. The elongation of the C-H distance that
also has to be expected has much less influence. So the planar moment per pair of hydrogens should get smaller
with increasing ring size. Taking into account the effect of out-of-plane vibrations on the planar moment - which
should be expected to be higher for six-membered rings than for five-membered rings, thus making the value of Pc
larger - this corresponds quite well with the experimental values.
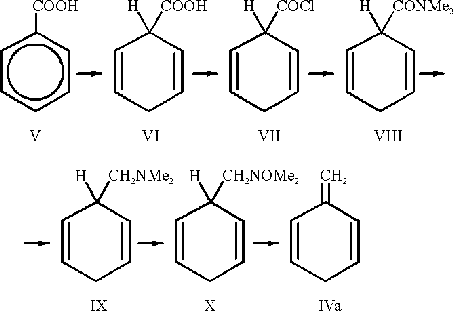
However a slight bending of the cyclohexadiene ring to form a very shallow boat cannot be ruled out by the planar
moment Pc alone. If the observed value is to be reproduced the dihedral angles within the ring can be as large as
about 3 degrees if the C-H bond length and the H-C-H angle are to remain within acceptable limits (cf. figure
below). Yet, a value of almost 8 degrees, as determined by Trætteberg et al. [4] for the 4,4-dimethylated species, is
not reconcilable with these results.
Dependence of the H-C4-H angle and the C4-H distance on variation of the dihedral angles (C6-C1-C2-C3 and C2-C3-C4-C5) in the ring.
The abscissa represents the H-C-H angle in degrees, the ordinate the C-H distance in Å. For nine dihedral angles (varying between 0 degrees and
8 degrees) the data points show the possible values of the methylene geometry to reproduce the experimental planar moment Pc. The dashed lines
denote extreme values between which the parameters should be expected to lie.
For a final decision it was therefore desirable to be able to calculate the actual contribution of the two hydrogens in
position 4 to the planar moment. This can be done if their coordinates (especially the c coordinates) are known. So
we decided to synthesize and measure 4-monodeuterated 1-methylene-2,5-cyclohexadiene.
Purity and degree of deuteration were checked by mass spectrometry and NMR. Both methods showed that the
degree of deuteration was well over 90%.
The rotational constants that were predicted by ab initio calculations for the parent molecule were compared to the
experimental data. The relative deviation for each constant was multiplied with the corresponding constant for the
deuterated molecule obtained from the ab initio geometry (cf. Table 4). The rotational
constants thus calculated predicted rotational transitions near 39 GHz (J=9) with deviations of less than 1 MHz. So
the spectrum of the deuterated compound could be assigned with no substantial difficulties. We measured 44
transitions between 28 and 40 GHz with a maximum J of 49 (cf. Table 5). The least squares fit showed a standard
deviation of 18 kHz and yielded the rotational constants as well as all five quartic centrifugal distortion constants
(see Table 2).
By a qualitative evaluation of the observed spectra first indications for a planar ring can be obtained. As Avirah et
al. [13] have pointed out there are several possibilities with respect to the potential function of the ring-bending
vibration (and thus to the planarity question):
A first possibility would be a double minimum potential with a high barrier (> 500 cm-1). In this case the
rotational spectrum in the ground state of the normal isotopomer would possibly show no indication of the barrier
(except the planar moment, which would be higher than expected). The 4-monodeuterated species, however, would
exist in two distinct conformers (flagpole or bowsprit deuterium) which would have different microwave spectra. It
cannot be definitely ruled out that only transitions of one of the conformers have been assigned and the others have
gone unnoticed, but the probability would be rather small, as the transitions that were predicted using rotational
constants calculated for a planar conformation were correct within 1 MHz. This would be very unlikely if the ring
were not planar. So this possibility can be ruled out.
A second possibility is a double minimum potential with a medium height barrier (~100-500 cm-1). In this case the
interaction between the two degenerate vibrational levels would become so strong that the splittings caused in the
microwave spectrum should make it impossible to fit the observed transitions to a planar model.
The third possibility is a double minimum potential with a barrier below the vibrational ground state energy. An
example of such a case is cyclobutanone [14]. Without investigation of the microwave spectrum in the excited
vibrational states such a case cannot be distinguished from the fourth possibility, a single minimum potential.
So the only potentials that would be consistent with the observed microwave spectra are either a single minimum
potential or a double minimum potential with a barrier below the vibrational ground state. In either case, however,
the vibrational ground state structure would have to be considered planar.

Deuteration
First we tried to deuterate the amine (IX), but it turned out that no significant exchange of deuterium for hydrogen
could be achieved. Therefore we prepared a p-deutero substituted benzoic acid
(XI) according to the following
reaction scheme: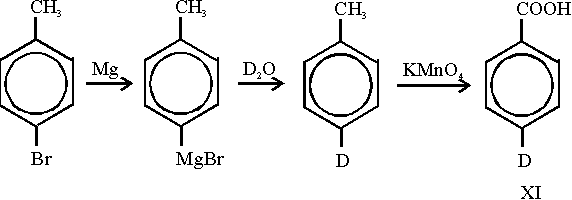 Starting from p-bromo-toluene deuteration was achieved via a Grignard compound which was treated with
deuterium oxide. The methyl group is easily oxidized by potassium permanganate. From the resulting 4-deutero-
benzoic acid (XI) the preparation of 4-deutero-1-methylene-2,5-cyclohexadiene
(IVc) was carried out as mentioned
above for the non-deuterated compound.
Starting from p-bromo-toluene deuteration was achieved via a Grignard compound which was treated with
deuterium oxide. The methyl group is easily oxidized by potassium permanganate. From the resulting 4-deutero-
benzoic acid (XI) the preparation of 4-deutero-1-methylene-2,5-cyclohexadiene
(IVc) was carried out as mentioned
above for the non-deuterated compound. Theoretical calculations
Just as for the analogous oxo-compound 4,4-dimethyl-2,5-cyclohexadienone all ab initio and all semi-empirical
calculations predicted a planar ring for methylene-cyclohexadiene. It is a well-known fact that particularly semi-
empirical methods (we used MINDO/3 [18], MNDO [19] and AM1 [20] with the software package MOPAC [21])
are too much in favor of planar rings. But ab initio calculations with GAUSSIAN86 [22] gave the same result with
both smaller (STO-3G) and larger (6-31G*) basis sets. Rotational constants, dipole moment and the geometry of
the ring methylene group were predicted remarkably well by a RHF-SCF ab initio calculation with a 6-31G* basis
set. A survey of some of the results is given in Table 6.
 2 1,643 [10]
2 1,643 [10]
 2 1,639 [11]
2 1,639 [11]
 2 1,699 [12]
2 1,699 [12]
 2 1,652 [13]
2 1,652 [13]
 3 1,663 [14]
3 1,663 [14]
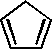 1 1,551 [15]
1 1,551 [15]
 1 1,523 [16]
1 1,523 [16]
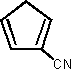 1 1,520 [16]
1 1,520 [16]
 1 1,531 (35Cl) [17]
1 1,583 (37Cl) [17]
1 1,531 (35Cl) [17]
1 1,583 (37Cl) [17]
 1 1,588 This work
_________________________________________________________________________
1 1,588 This work
_________________________________________________________________________  A 3338.80 3276.33 3260.03 3306.67 3370.32 3332.16
B 1205.23 1178.95 1165.69 1151.86 1197.24 1193.07
C 1092.49 1067.37 1061.41 1050.04 1084.41 1082.21
m 4.8 2.9 3.6 4.3 4.2 4.45
___________________________________________________________________________
A 3338.80 3276.33 3260.03 3306.67 3370.32 3332.16
B 1205.23 1178.95 1165.69 1151.86 1197.24 1193.07
C 1092.49 1067.37 1061.41 1050.04 1084.41 1082.21
m 4.8 2.9 3.6 4.3 4.2 4.45
___________________________________________________________________________
 A 5194.16 5100.61 5167.73 5181.18 5254.50 5177.82
B 2645.64 2634.15 2559.82 2523.76 2621.14 2613.15
C 1771.38 1755.56 1730.68 1714.79 1768.96 1755.84
|a| 2.538 2.543 2.567 2.600 2.541 2.555
|c| 0.865 0.872 0.892 0.873 0.904 0.867
m 0.8 0.6 0.1 0.1 0.5 0.86
A 5194.16 5100.61 5167.73 5181.18 5254.50 5177.82
B 2645.64 2634.15 2559.82 2523.76 2621.14 2613.15
C 1771.38 1755.56 1730.68 1714.79 1768.96 1755.84
|a| 2.538 2.543 2.567 2.600 2.541 2.555
|c| 0.865 0.872 0.892 0.873 0.904 0.867
m 0.8 0.6 0.1 0.1 0.5 0.86